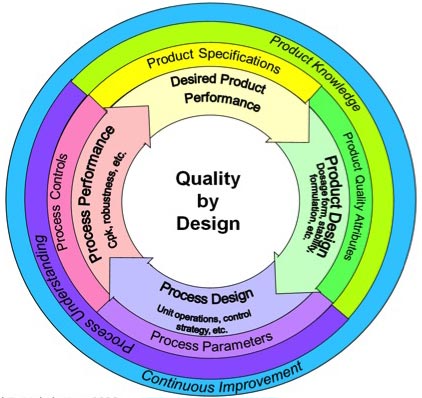
Diário
de bordo: O controle estatístico de
processo (CEP) consiste na aplicação de gráficos estatísticos para verificação
da qualidade da fabricação detectando desvios de parâmetros representativos do
processo com a finalidade de reduzir a quantidade de produtos fora de
especificações e com isso os custos da produção. Embora não pareça, a
utilização do CEP na indústria farmacêutica ainda é incipiente, pois a abordagem
do controle durante o processo prevalente em muitas empresas tem como base a
mera comparação dos resultados com metas arbitrárias levando-se em conta a
especificação do produto. Este procedimento, embora indique onde o processo se
encontra, não é capaz de demonstrar como o processo chegou lá, não revelando
qualquer compreensão de como o processo funciona, ignorando a variação dentro
do processo e tratando cada flutuação apenas como uma sinalização de correção
não agindo sobre a verdadeira causa. O CEP apresenta-se como uma ferramenta que
incorpora o conceito de Boas Práticas de Fabricação (BPF) e, por permitirem a
investigação dos pontos críticos de controle, fornece informações
indispensáveis para a validação de processos, diagnosticando possíveis não
conformidades em todas as etapas do processo, além de sinalizar as possíveis
fontes de desvios de qualidade possibilitando correções e interações com o
processo.
AS BOAS PRÁTICAS DE FABRICAÇÃO (BPF)
As BPF nas indústrias farmacêuticas que possui como
base garantir a qualidade do produto farmacêutico tendo-se em vista o risco
sanitário através de monitoramentos específicos em todas as etapas da produção,
também incorporam os conceitos de otimização de processos, redução de perdas e
gestão ambiental. Para haver esta sincronização entre estes conceitos, cada vez
mais, as atividades de controle do processo devem interagir com as demais operações,
controles de fabricação e aplicações de gerenciamento.
A Agência Nacional de Vigilância Sanitária (ANVISA)
adota a garantia de qualidade para o
Medicamento, que consiste no acompanhamento de todo
o processo desde a aquisição de uma matéria-prima farmacêutica pelo fabricante
até sua transformação em um produto acabado à disposição do consumidor. Para tanto publicou uma série de
regulamentações a todas as etapas da cadeia do medicamento e executa ações de
fiscalização para avaliar a qualidade dos processos produtivos de fabricação, das
condições de armazenagem, transporte e consumo desses produtos.
Avaliando este cenário, cuja intenção é a busca por
melhoria da qualidade, os fabricantes devem concentrar seus esforços em
melhoramentos contínuos, ou seja, implementando dispositivos que permitam
reconhecer problemas, priorizar ações corretivas, implementá-las e manter uma
atitude proativa com relação aos
procedimentos operacionais padronizados.
A utilização de métodos estatísticos não garante a solução
de todos os problemas de um processo, porém é uma maneira racional, lógica e
organizada de determinar onde eles existem, sua extensão e a forma de
solucioná-los. Esses métodos podem ajudar na obtenção de sistemas que assegurem
uma melhoria contínua da qualidade e da produtividade ao mesmo tempo.
O CONTROLE ESTATÍSTICO DO PROCESSO (CEP)
O CEP consiste em um conjunto de ferramentas de
monitoramento on-line da qualidade.
Através destas ferramentas obtém-se uma visão detalhada do comportamento do
processo, identificando variações e possibilitando seu controle ao longo do
tempo. Isto se dá por meio de continua coleta de dados, análise e bloqueio de
prováveis causas especiais, responsáveis por instabilidades no processo avaliado.
O principal objetivo do CEP é o melhoramento dos processos produtivos
conferindo-lhes menor variabilidade e proporcionando-lhes melhores níveis de
qualidade nos resultados da produção.
A estratégia na aplicação do CEP é: se se conhece um
determinado processo pode-se prever toda a sua ocorrência, ou seja, se um
processo ocorre sob condições conhecidas e estas são cuidadosamente mantidas,
este processo estará sujeito apenas aos efeitos de causas comuns os quais
definem a posição e a dispersão deste processo. Estatisticamente falando isto
representa uma Distribuição Normal. Desta maneira, agir no processo significa
evitar defeitos independentemente de onde eles possam manifestar-se. Este é o
principio do CEP: agir sobre o processo e não no produto utilizando da
estatística como instrumento para organizar, tratar e analisar as informações
do processo. Assim o CEP opera preventivamente utilizando-se de uma base
objetiva de análise e atuando de forma abrangente não se limitando a casos
específicos, mas na produção como um todo e finalmente permitindo uma adequada
avaliação da qualidade.
O CEP assim disposto constitui-se numa ferramenta
útil tanto na melhoria como na validação dos processos farmacêuticos podendo
ser aplicado ao processo como um todo ou em suas etapas. O CEP permite
demonstrar estatisticamente que os processos podem ser compreendidos e assim
identificar e corrigir as causas das não conformidades que possam apresentar. A
identificação e correção trazem maior conhecimento e controle sobre os pontos
críticos do processo de fabricação. É importante assinalar que o CEP por si só
não determina a validação de um processo e que o estudo isolado de um único
parâmetro demonstrado no controle estatístico do processo (tais como:
desintegração, dureza, friabilidade, dissolução, peso médio, etc.) não pode ser
considerado uma validação.
Assim sendo o CEP apresenta-se como uma ferramenta de
aplicação simples, capaz de permitir maior compreensão do processo,
possibilitando ações rápidas de controle pelo pessoal de operação, rica nos
resultados que podem apresentar sobre o comportamento do processo e apta a ser
aplicada a vários processos farmacêuticos. Por ser uma ferramenta que oferece todos estes
recursos o CEP não deve prestar-se a ser um mero indicador de não conformidades.
O profundo conhecimento do processo que o CEP proporciona pode apontar de forma precisa quais os pontos críticos e
onde as melhorias dever ser realizadas tornando a produção robusta e um
procedimento seguro.
Embora o CEP apresente todo este potencial para a
gestão dos processos, a sua utilização só surtirá efeito com a consolidação da
cultura do pensamento estatístico em todos os níveis da empresa. Atualmente a indústria
farmacêutica abre-se para novas tecnologias, flexibilização de práticas
regulatórias, utilização de técnicas de modelagem e simulação de processos,
utilização de métodos quimiométricos nos seus processos e assumindo novas
práticas de gerenciamento. Todas estas tendências serão bem sucedidas se somente
se as empresas tiverem como base o CEP como ferramenta em sua prática
rotineira.
Texto baseado no artigo “Aplicação do controle
estatístico de processo na indústria farmacêutica” de Lima, A.A.N.; Lima,
J.R.; Silva, J.L.; Alencar, J.R.B.; Soares-Sobrinho, J.L.; Lima, L.G.;
Rolim-Neto, P.J. Publicado na Revista
de Ciências Farmacêuticas Básica e Aplicada, v. 27, n.3, p.177-187, 2006.
.jpg)