DIÁRIO DE BORDO: A
qualidade para produto farmacêutico é medida pela sua adequação ao uso
terapêutico, pela sua especificação e atributos e seu acesso tanto no que se
refere tanto à sua distribuição quanto pelo seu custo. E uma característica
básica é que a o aspecto segurança e o aspecto qualidade não podem ser
separáveis por causa do risco sanitário embutido na sua utilização. Desta
preocupação surgiram as Boas Práticas de Fabricação, um código legal dos
princípios de qualidade aplicados na fabricação e controle de produtos
farmacêuticos cujo objetivo é garantir que as matérias-primas, utilizadas na
fabricação dos medicamentos sejam padronizadas e livres de contaminações; o
processo de fabricação seja capaz de produzir um produto farmacêutico dentro
dos atributos de qualidade previstos e que medições e ensaios adequados foram
utilizados para assegurar as especificações de qualidade até o final do seu
prazo de validade. Desta maneira os fabricantes podem perder alguns lotes
até as reais causas de falha ser compreendidas e solucionadas. Tais
especificações rigorosas podem resultar em recolhimentos de produto e falta de
medicamentos no mercado. Recursos significativos são desperdiçados debatendo-se
problemas relacionados à aceitação da variação, necessidade de testes adicionais
de controle, e estabelecimento de critérios de aceitação. Requisitos
regulatórios impõem notificações para a execução de mudanças mínimas e
incrementais nos processos e controles inibindo assim melhorias contínuas e
estratégias para implantação da garantia da qualidade em tempo real.
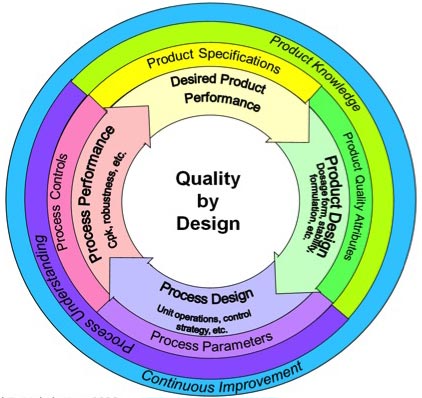
O QbD surge desta
necessidade de maior flexibilização nos processos de fabricação, pois
apresenta-se com uma abordagem proativa, sistemática, científica, com base em
análise de risco do desenvolvimento farmacêutico. Começa com objetivos
pré-definidos e com destaque nas concepções do produto, do processo e do
controle. Isto significa planejar e desenvolver formulações e processos de
manufatura que garantam os objetivos pré-definidos da qualidade do produto
através da identificação das características críticas de qualidade do ponto de
vista do paciente, traduzindo-se em atributos que o produto deve apresentar
independentemente das variações dos parâmetros críticos do processo. Para fazer
isto as relações entre as variáveis da formulação e do processo produtivo
(incluindo os atributos dos insumos ativos e dos excipientes, assim como os
parâmetros do processo) e as características do produto são estabelecidas e as
fontes de variação identificadas. O conhecimento assim adquirido é utilizado
para implementar um robusto e flexível processo de manufatura que pode se
ajustar e produzir um produto consistente repetidamente.
No modelo QbD, a
qualidade do medicamento é assegurada pelo entendimento e controle das
variáveis de formulação e de manufatura. Os ensaios analíticos aplicados ao
produto terminado apenas confirmam a sua qualidade. No modelo atual a
especificação é frequentemente fixada pela observação de dados de um pequeno
número de lotes que fixam os critérios de aceitação para os futuros lotes a
serem produzidos. No modelo QbD a consistência vem do planejamento e controle
do processo produtivo e da especificação do medicamento.
O
termo “Quality by Design” foi criado na década de 60 por Joseph M. Juran um
engenheiro que acreditava que a maioria dos problemas referentes à qualidade de
um produto está relacionada com o modo como o desenvolvimento deste produto foi
realizado e assim sendo quanto mais sistemáticos os estudos para a definição e
adequação do uso pretendido e, quanto mais detalhadas as características que
garantem a sua eficácia durante a fase de desenvolvimento deste produto, quanto
melhor o seu desempenho na fase produção e comercialização. Portanto,
para Juran o produto deve ser concebido e construído levando-se em
consideração a qualidade, a segurança e efetividade; a qualidade não
pode ser testada no produto; e cada etapa do processo deve ser controlada para
maximizar a probabilidade de que o produto final alcance seus atributos de
qualidade e as especificações (“Quality Control Handbook” -1950)
O QbD
foi proposto primeiramente pela agencia reguladora dos EUA ( FDA- a partir de
2002) e seguida pela agencia reguladora da comunidade europeia (EMA) e pela
agencia reguladora do Japão (PMDA). O processo está sendo conduzido pelo ICH
(International Conference on Harmonization) entidade em que participam grupos
destas três agencias reguladoras. O ICH já desenvolveu três guias que orientam
para os conceitos de QbD, a saber:
• ICH-Q8 Desenvolvimento
Farmacêutico (2004)
• ICH-Q9
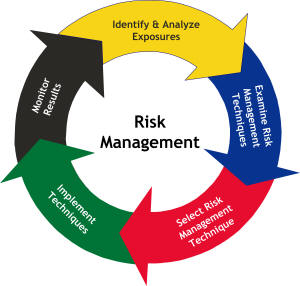
Gerenciamento do Risco da Qualidade (2006)
• ICH-Q10
Sistema Farmacêutico da Qualidade (2007)
Estas
guias começam a ser internalizadas nas agencias reguladoras dos EUA e EMA a
partir de 2012.
No
Brasil alguns conceitos começam a ser incentivados pela ANVISA e praticados
pelos fabricantes, tais como o gerenciamento de risco, o incremento do controle de mudanças e o monitoramento
periódico do desempenho do produto durante todo o ciclo de vida do produto.